Research Overview
Our work focuses on the intracellular life cycles of two human pathogens: Rickettsia parkeri and Listeria monocytogenes. R. parkeri is an obligate intracellular Gram-negative bacterium that causes a mild spotted fever disease and is transmitted to humans by ticks. In contrast, L. monocytogenes is a facultative Gram-positive bacterium that cause listeriosis after ingestion of contaminated food sources. Both pathogens invade cells, enter the cytosol, and hijack host actin to promote actin-based motility. Motile bacteria then initiate cell-to-cell spread, a step-wise process during which bacteria propel themselves to the host cell membrane, form double-membrane protrusions into the recipient cell, and then become engulfed into a vesicle before escape to the recipient cell cytosol. These intricate, complex life cycles illustrate the different ways pathogens interact with their host and we are interested in elucidating the cellular and molecular mechanisms responsible. We investigate these mechanisms using an interdisciplinary approach combining cell biology, microbiology, genetics, biochemistry, and biophysics. We also engineer tools capable of probing the host and bacterial factors that regulate pathogenesis. In the end, our goals are to reveal important properties of pathogenesis and to use pathogens as tools to better understand host cell biology.
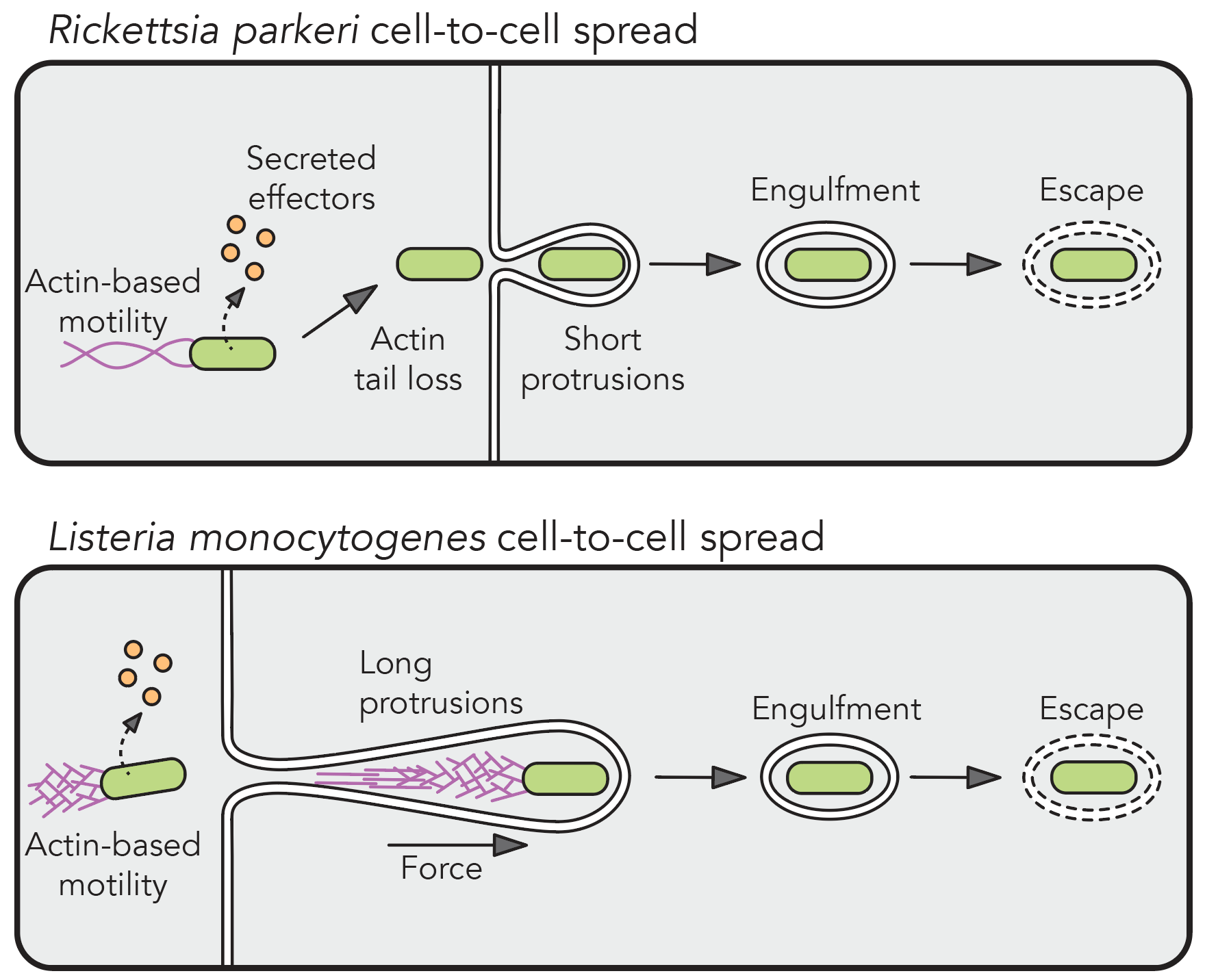
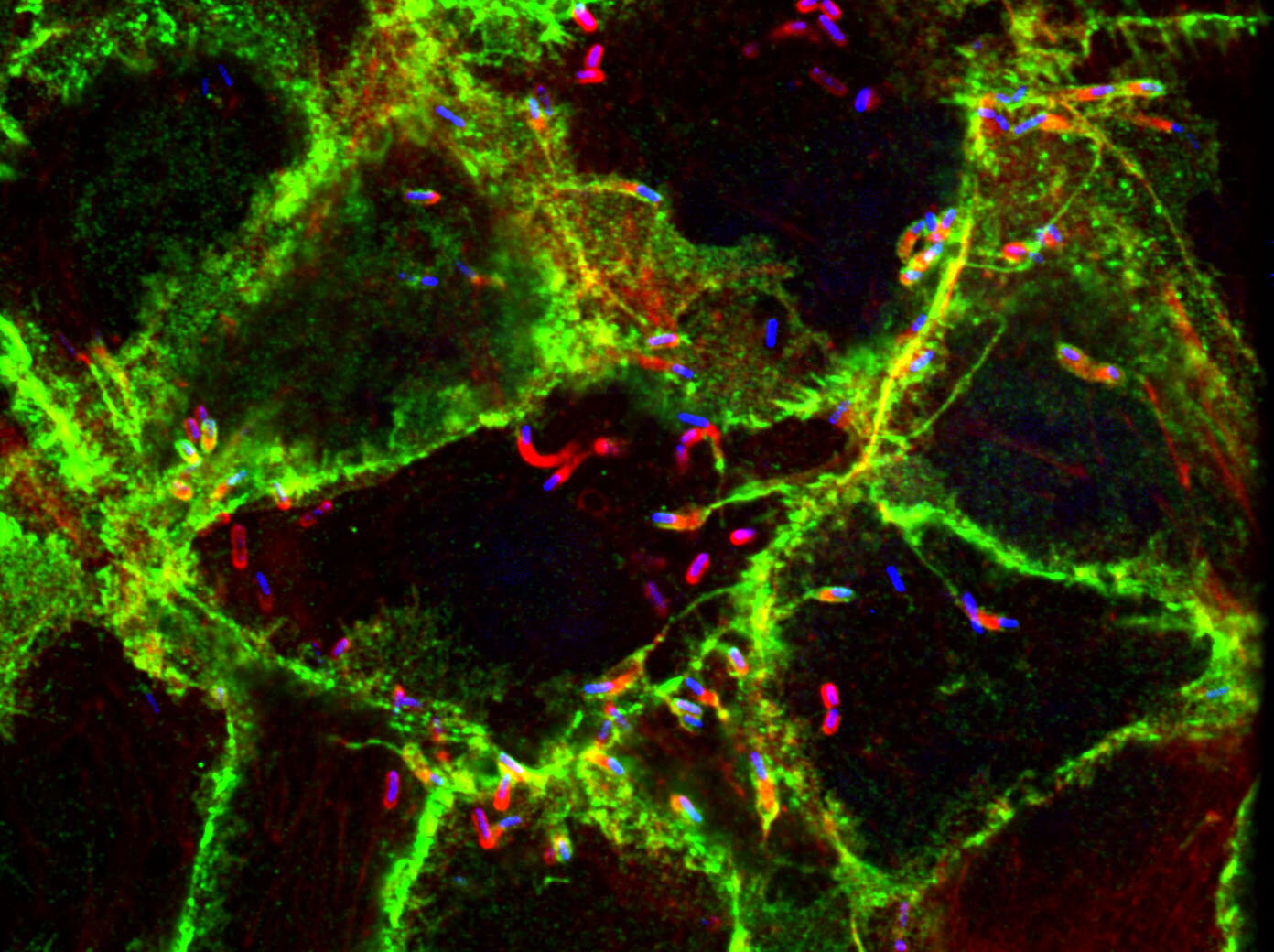
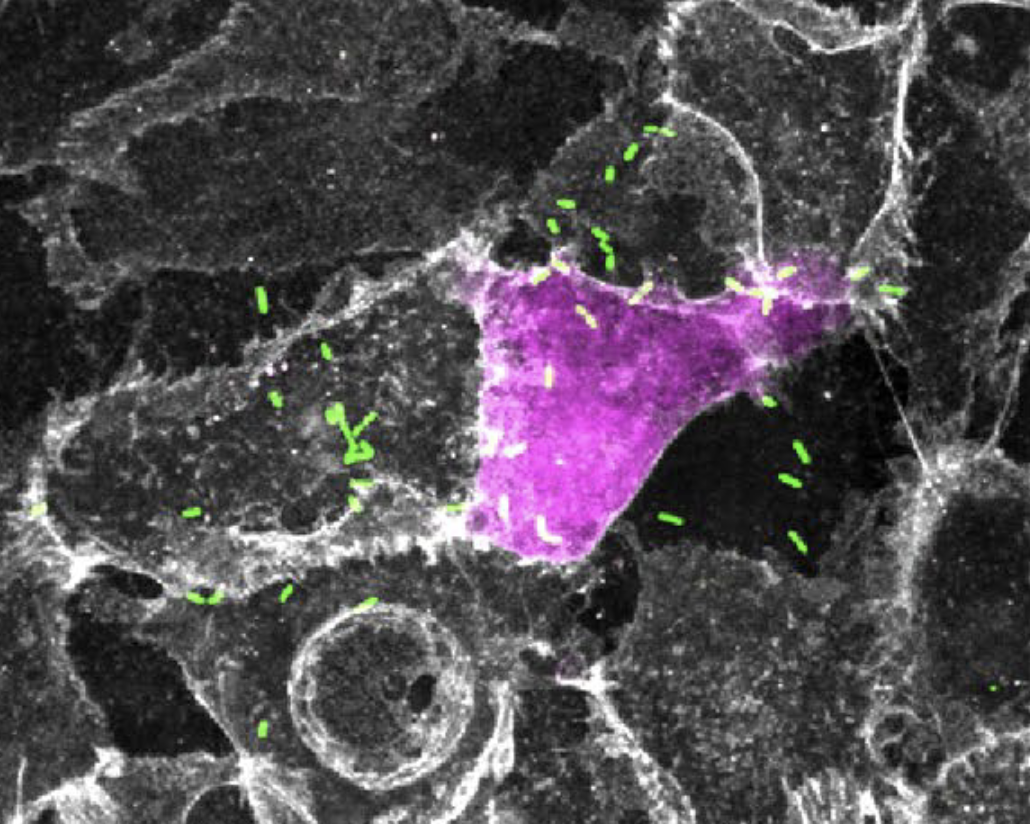
Cell-to-cell spread
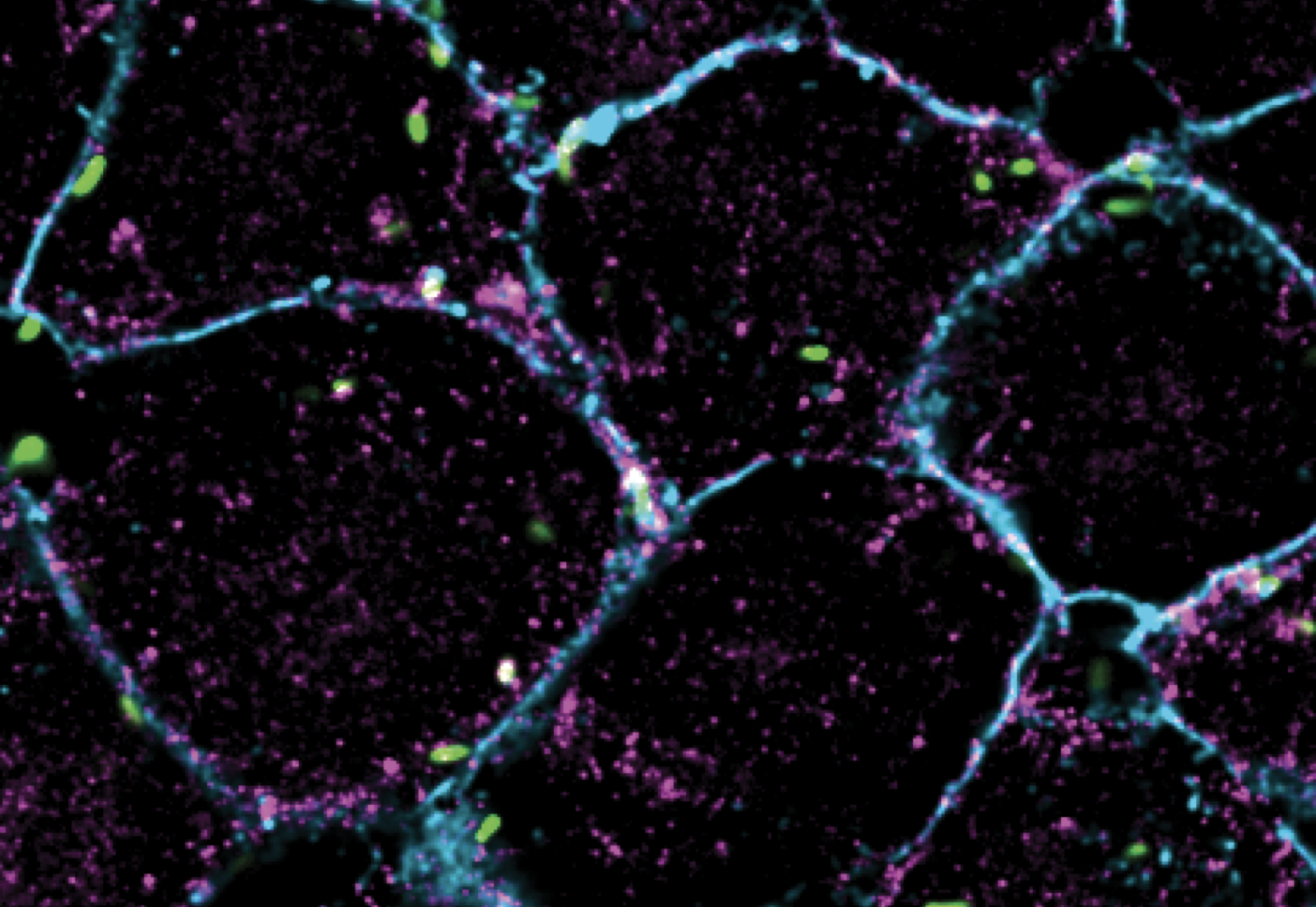
Cell-to-cell spread is a critical yet understudied virulence mechanism that R. parkeri and L. monocytogenes use to promote tissue-wide dissemination. During cell-to-cell spread, bacteria remodel host cell-cell junctions into double-membrane protrusions that are engulfed by neighboring host cells. Intriguingly, these pathogens do not use the same strategies for spread, and we are investigating how these different mechanisms are regulated by examining the critical host and bacterial factors involved.
The study of cell-to-cell spread also allows us to ask questions about how host cell-cell junctions are remodeled by specific host pathways. Leveraging our microbial tools for cell biological studies, we aim to understand how mechanoresponsive, trafficking, and adhesion machinery respond to the dramatic shape and force changes during the process of bacterial cell-to-cell spread, which may shed light on how cells regulate the intercellular movement of other large cargo like organelles.


INTERACTIONS between Pathogens & Organelles
While much of our work has focused on interactions with the plasma membrane during invasion and spread, we are discovering additional interactions between bacterial pathogens and the membranes within our cells. We explore this using a combination of different imaging modalities (confocal, electron microscopy, FIB-SEM), and we hope to uncover the molecular mechanisms driving these interactions and their functional consequences for the host, pathogen, or both.
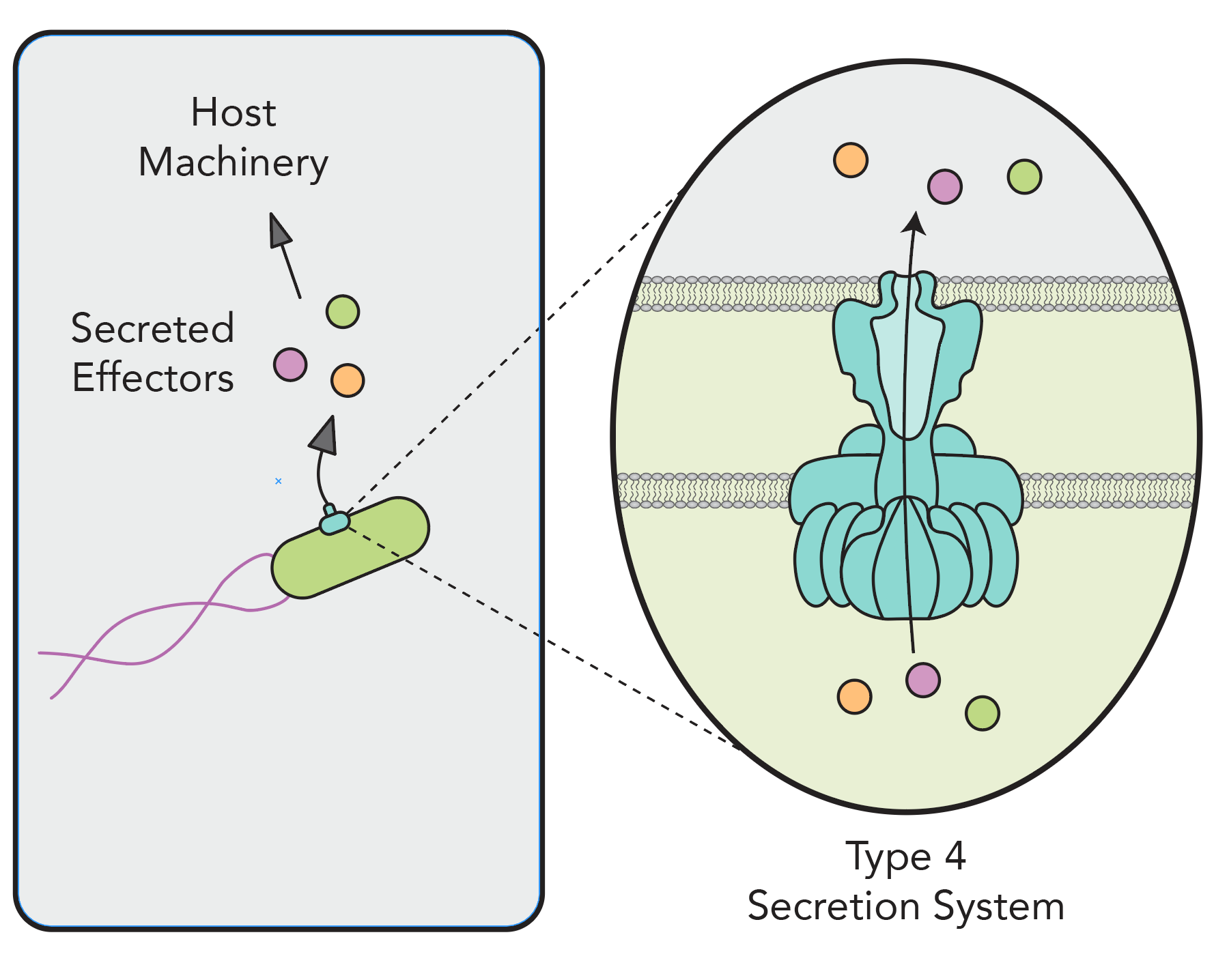
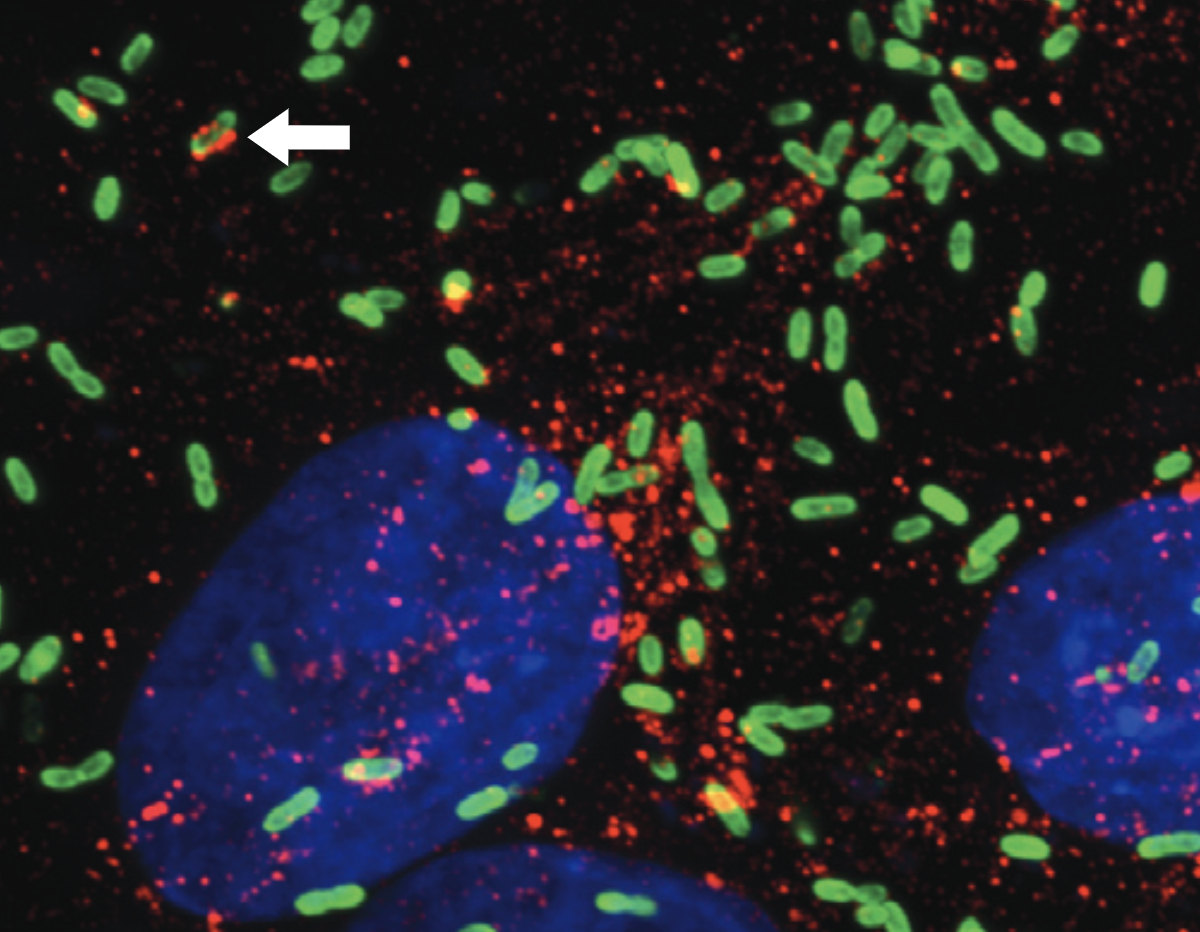
SECRETED EFFECTORS AND SECRETION Systems
Bacterial pathogens deploy proteins called secreted effectors into host cells that hijack host cell processes to promote infection. Notably, very little is known about the identity and function of the Rickettsia secretome or the machines responsible for secretion. We are combining our established functional-genetic approaches with biochemical tools like BONCAT to identify and study the role of secreted effectors during Rickettsia infection. We are also using structural biology to investigate how an anomalous Type 4 Secretion System assembles to drive effector delivery during infection.
Expanding the toolkit for studying host-pathogen interactions
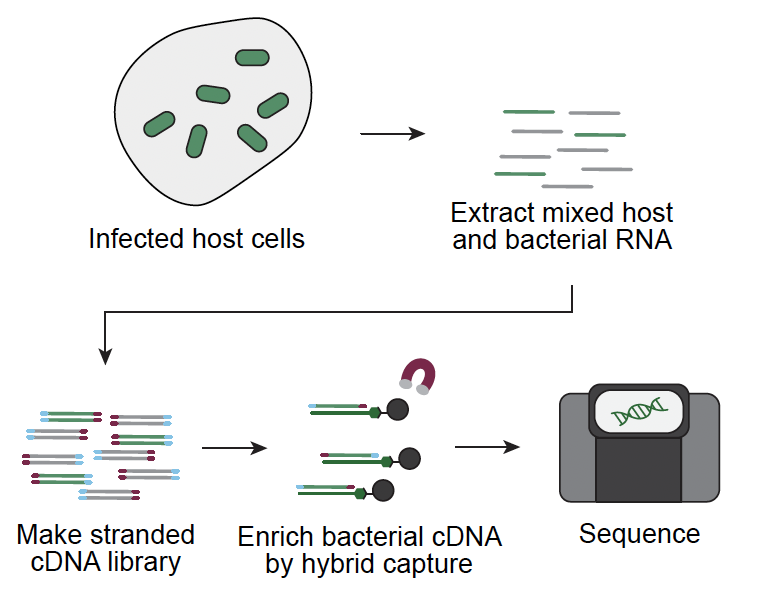
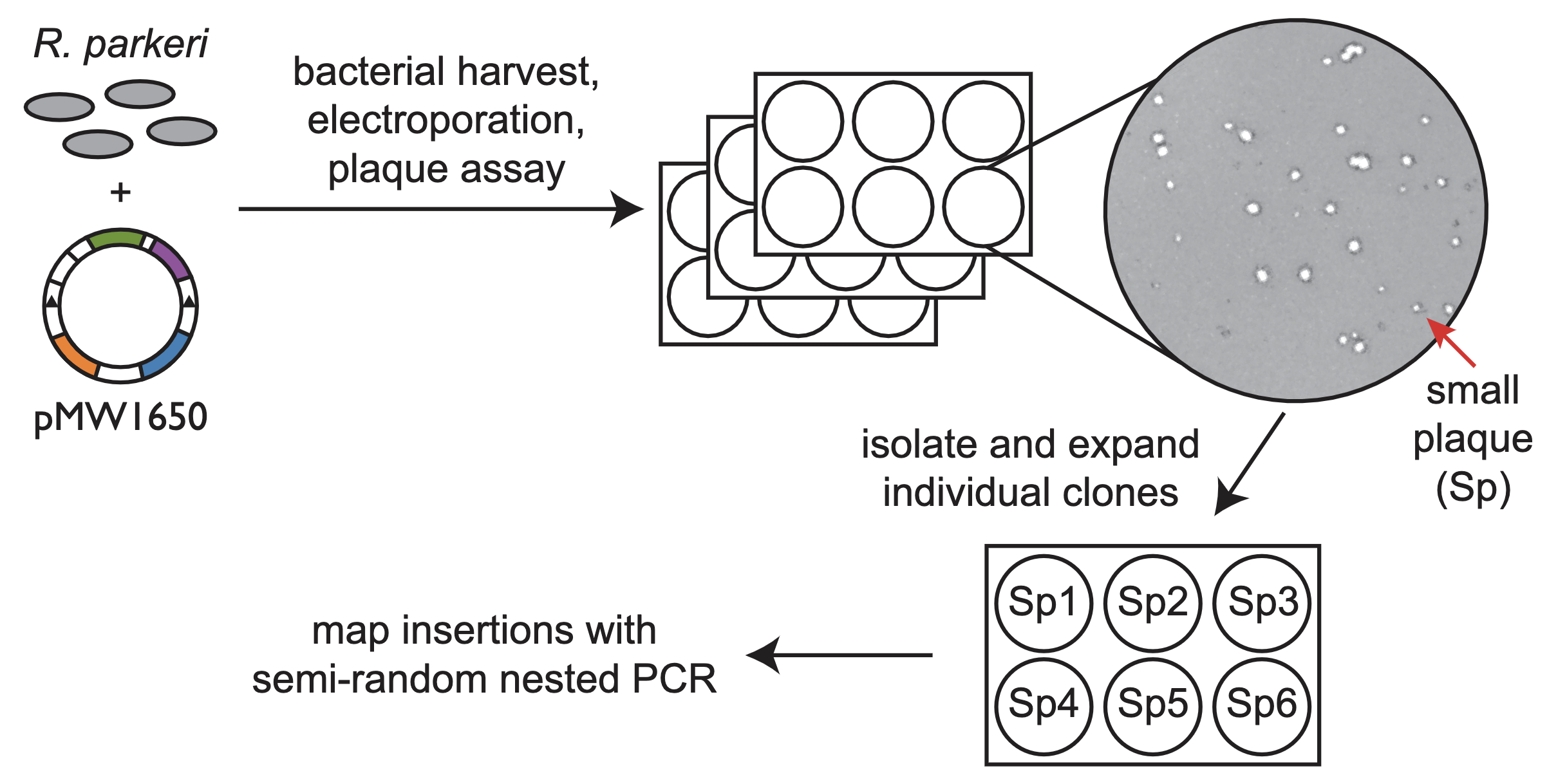
We have pioneered several key advances in the fields of Rickettsia and Listeria pathogenesis, including advanced proteomic profiling, improved genetic manipulation and mutagenesis, and more sensitive RNA-seq. Our goals are to 1) use these tools to accelerate our understanding of the factors regulating infection and 2) share these tools with the broader community to motivate collaborative innovations in technology development.


-
Scott AT, McGinn J, Butty V, Levine SS, Lamason RL. Hybrid capture RNA-seq defines temporal gene expression in Rickettsia 2026 preprint bioRxiv
-
-
Sanderlin AG, Margolis HK, Meyer AF, Lamason RL. Cell-selective proteomics reveal novel effectors secreted by an obligate intracellular bacterial pathogen. 2024. Nature Communications. 15, 6073 Article
Woida PJ, Smith MW, Lamason RL. Proximity labeling of the Listeria monocytogenes surface reveals pathogen control of a host deubiquitinase. 2026 preprint
bioRxiv -